
Sinker” Syndrome in Equine Laminitis: Pathophysiology, Metabolic Changes and Biomechanical Catastrophe
Laminitis remains one of the most devastating conditions in equine medicine, characterised by failure of the suspensory apparatus of the distal phalanx (SADP) and subsequent displacement of the distal phalanx (P3) within the hoof capsule. Among the most catastrophic presentations is “sinker” syndrome, defined by circumferential distal displacement of the distal phalanx with compression of the coronary corium and complete collapse of the laminar suspensory mechanism (Pollitt, 2004; Patterson-Kane & Karikoski, 2018). Unlike rotational or focal displacement of P3, sinker syndrome represents global failure of the laminar interface, often progressing rapidly and with a grave prognosis. This essay explores the pathophysiology of sinker syndrome, with particular focus on the metabolic drivers and biomechanical sequelae that render this condition catastrophic.
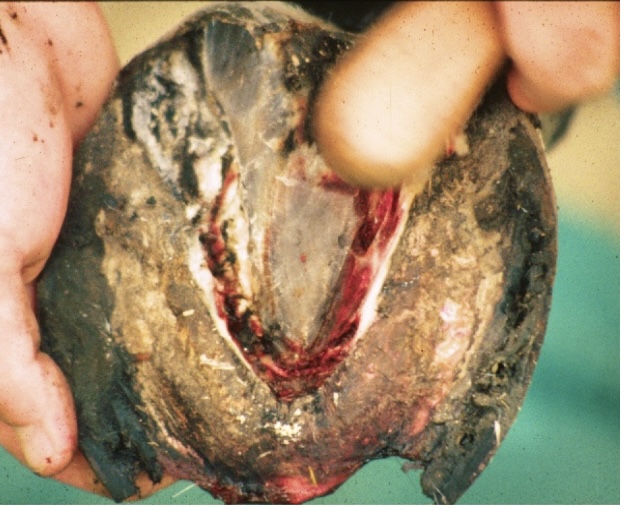
Pathophysiology of Sinker Syndrome
Metabolic and Inflammatory Basis
The initiating events in laminitis are increasingly recognised as systemic, with endocrine and inflammatory derangements central to laminar failure (Belknap & Black, 2012). In endocrine laminitis, hyperinsulinaemia plays a pivotal role. Sustained high circulating insulin concentrations stimulate abnormal insulin-like growth factor-1 receptor signalling in laminar basal epithelial cells, driving cytoskeletal dysregulation and weakening adhesion to the basement membrane (de Laat et al., 2013). Inflammatory mediators—particularly matrix metalloproteinases (MMPs), cytokines, and reactive oxygen species—are upregulated during sepsis-associated laminitis, contributing to enzymatic degradation of the extracellular matrix (Loftus et al., 2007).
In sinker syndrome, this biochemical disruption is not confined to localised laminar segments but extends circumferentially, leading to global detachment of P3. Loss of the interdigitation between epidermal and dermal lamellae eliminates the tensile strength normally resisting the constant ground reaction forces transmitted through the hoof wall (Pollitt & Collins, 2010).
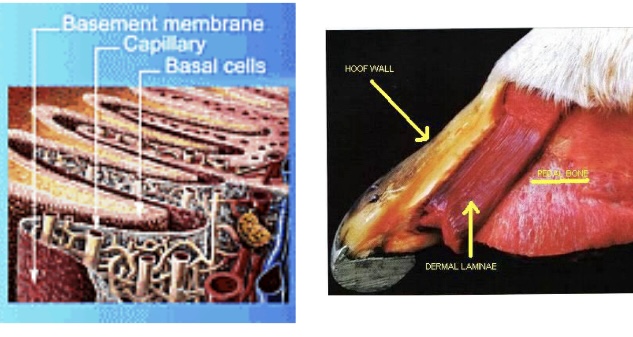
Vascular Dysregulation
A hallmark of sinker progression is vascular collapse within the coronary corium and laminar bed. Endothelial dysfunction—driven by endotoxaemia, platelet activation, and altered nitric oxide signalling—precipitates vasoconstriction, capillary congestion, and ischaemia (Hood, 1999). This leads to impaired perfusion of the laminar tissues, compounding the metabolic compromise and hastening necrosis. Compression of the coronary band vessels as P3 descends exacerbates the ischaemia, resulting in visible coronary sinking and bruising of the dorsal coronary margin (Patterson-Kane & Karikoski, 2018).
Biomechanical Catastrophe
Disruption of the Suspensory Apparatus of P3
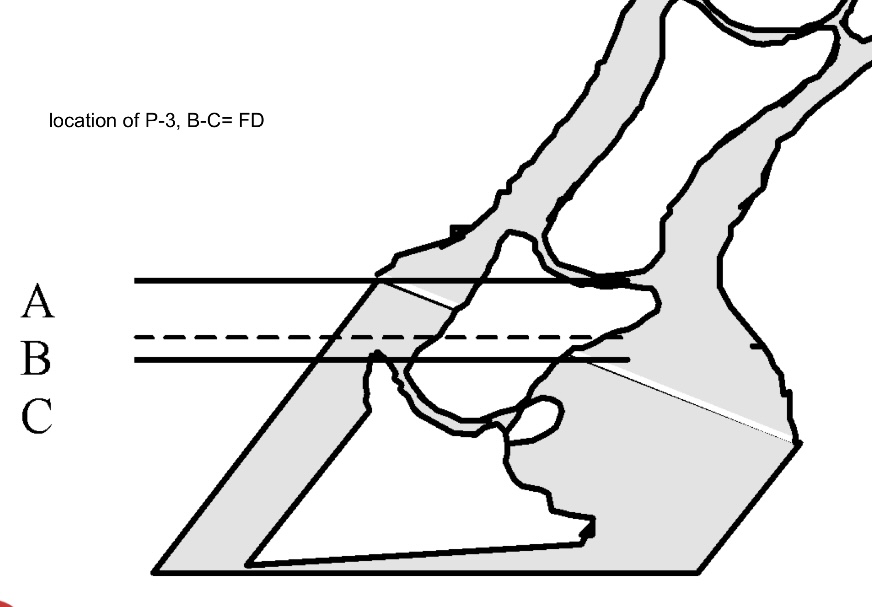
The hoof-laminar apparatus normally suspends P3 within the hoof capsule, distributing forces evenly across thousands of primary and secondary lamellae. In sinker syndrome, complete detachment of these lamellae removes the suspension system, allowing gravity and the constant ground reaction force to drive P3 distally (Thompson et al., 2008). The circumferential failure leads to uniform distal displacement rather than rotation about the dorsal lamellae.
Coronary Band and Sole Compression
As the distal phalanx sinks, the coronary corium becomes compressed between the proximal hoof wall and P3. This compromises coronary papillae function and horn production, leading to cessation of wall growth and severe pain at the coronary band (Pollitt, 2004). Simultaneously, the sole corium is crushed by the descending apex of P3, producing solar prolapse, bruising, and penetration in advanced cases (Curtis, 2020). These changes represent a vicious cycle of structural collapse, progressive tissue necrosis, and mechanical overload.
Altered Load Distribution and Secondary Pathology
Biomechanically, sinker syndrome disrupts the normal force distribution across the foot. With loss of laminar suspension, axial load transfers disproportionately to the sole and bars, creating high focal pressures that are poorly tolerated by the sole corium (Dyson, 2011). Horses often adopt a “walking on eggshells” gait, reflecting both pain and loss of mechanical integrity. Secondary complications include abscessation, sepsis from sole penetration, and destabilisation of the distal interphalangeal joint due to altered alignment (Hood, 1999).

Clinical Implications and Prognosis
Sinker syndrome is widely considered catastrophic because of its rapid progression, intractable pain, and limited treatment options. Radiographic signs include uniform distal displacement of P3, narrowing of the coronary band, and increased sole penetration risk (Patterson-Kane & Karikoski, 2018). Prognosis is grave, with many affected horses euthanised due to uncontrollable pain and systemic compromise. Therapeutic strategies such as deep bedding, sole support, cryotherapy, and mechanical unloading can provide temporary relief but rarely restore function once full circumferential detachment has occurred (Curtis, 2020).
Conclusion
Sinker syndrome represents the most severe biomechanical and metabolic manifestation of laminitis. Systemic endocrine and inflammatory drivers initiate laminar weakening, compounded by vascular dysregulation and enzymatic tissue degradation. Biomechanically, the collapse of the suspensory apparatus of P3 permits catastrophic distal displacement, crushing of the coronary and sole corium, and irreversible loss of hoof capsule function. The syndrome exemplifies how metabolic pathology and mechanical overload converge to produce one of the most devastating equine conditions, emphasising the importance of early prevention, rapid intervention, and recognition of its grave prognosis.
References
Belknap, J.K. & Black, S.J. (2012) ‘Equine laminitis: current concepts’, Equine Veterinary Journal, 44(6), pp. 738–744.
Curtis, S. (2020) Farriery: The Welfare and Therapeutic Approach. London: CRC Press.
de Laat, M.A., van Eps, A.W. & Pollitt, C.C. (2013) ‘Hyperinsulinaemia induces laminar pathology in horses’, Equine Veterinary Journal, 45(6), pp. 749–754.
Dyson, S.J. (2011) Diagnosis and Management of Lameness in the Horse. 2nd edn. St. Louis: Elsevier Saunders.
Hood, D.M. (1999) ‘The pathophysiology of developmental and chronic laminitis’, Veterinary Clinics of North America: Equine Practice, 15(2), pp. 321–343.
Loftus, J.P., Black, S.J. & Belknap, J.K. (2007) ‘Matrix metalloproteinase activity and laminitis: a review’, Veterinary Immunology and Immunopathology, 118(3–4), pp. 225–239.
Patterson-Kane, J.C. & Karikoski, N.P. (2018) ‘Laminitis: Recent advances and future perspectives’, Equine Veterinary Journal, 50(6), pp. 701–709.
Pollitt, C.C. (2004) Equine Laminitis. Lexington: International Veterinary Information Service.
Pollitt, C.C. & Collins, S.N. (2010) ‘The anatomy and physiology of the suspensory apparatus of the distal phalanx’, Veterinary Clinics of North America: Equine Practice, 26(1), pp. 29–49.
Thompson, K.N., Hood, D.M. & Wagner, I.P. (2008) ‘Pathophysiology of laminitis’, in Ross, M.W. & Dyson, S.J. (eds.) Diagnosis and Management of Lameness in the Horse. St. Louis: Elsevier Saunders, pp. 328–335.